The objective of this paper was to develop and validate new classification criteria for adult and juvenile idiopathic inflammatory myopathies (IIM) and their major subgroups. More specifically, the goal was to define minimum essential, easily available clinical and laboratory features to 1) distinguish IIM from similar but different conditions with high sensitivity and specificity, and 2) distinguish the major subgroups of IIM. The researchers used criteria from published documents and expert opinion and employed statistical analysis to identify criteria and consensus methodology to arrive at their conclusions. Consensus methodology is used to establish agreement on controversial subjects through a structured process.
The paper describes the Idiopathic Inflammatory Myopathies (IIM), which are collectively known as myositis, as typically exhibited by muscle weakness and muscle inflammation. The common subgroups are dermatomyositis (DM), polymyositis (PM), and inclusion body myositis (IBM) in adults; and juvenile dermatomyositis (JDM) in children.
Clinical trials and studies require patient groups in clearly defined categories, but the multiple diagnostic and classification criteria used for IIM are problematic. For outcome measures and improvement definitions in clinical trials, the International Myositis Assessment and Clinical Studies (IMACS) Group has reached consensus. But for the diagnostic and classification criteria, many are not validated and have significant limitations. The Bohan and Peter criteria are the most used, but they are not explicitly defined and do not specify how to exclude other forms of disease, leading to potential misclassification. Recent research has identified additional, potentially valuable features, such as myositis-specific autoantibodies, but they have not been tested adequately.
In 2004, the International Myositis Classification Criteria Project (IMCCP) was formed as an international collaboration with experts from adult and pediatric rheumatology, neurology, dermatology, epidemiology and biostatics. The IMCCP formed a steering committee and a working committee which gathered data for inclusion in the study. The study included data from 976 IIM patients (74.5% adults, 25.5% children) that was collected between 2008 and 2011 from 47 sites across the world. The subjects included the following cases:
| Subgroup | Number of Cases |
| Juvenile Dermatomyositis (JDM) | 248 |
| Polymyositis (PM) | 245 |
| Dermatomyositis (DM) | 239 |
| Inclusion Body Myositis (IBM) | 176 |
| Amyopathic Dermatomyositis (ADM) | 44 |
| Hypomyopathic Dermatomyositis | 12 |
| Immune-Mediated Necrotizing Myopathy (IMNM) | 11 |
| Juvenile Polymyositis | 1 |
Additionally, to ensure specificity and sensitivity in the criteria, the study included 624 comparison cases (81.6% adults, 18.4% children) with conditions that can mimic IIM. These cases included the following conditions:
- Systemic inflammatory diseases (36.5%)
- Muscle dystrophies (16%)
- Drug-associated or toxin-associated myopathies (7.9%)
- Motor neuron diseases/neuropathies (7.7%)
- Metabolic myopathies (6.9%)
- Myalgias (4.5%)
- Dermatological diseases (3.7%)
- Endocrine myopathies (3.7%)
- Infectious myopathies (4.5%)
- Mitochondrial myopathies (2.4%)
- Neuromuscular diseases (2.6%)
- Other myopathies (1.9%)
- Immune-mediated skin conditions (0.5%)
- Other diagnoses (1.3%)
The study used a probability-score model which added score points assigned to signs and symptoms and applied statistical analysis to predict IIM. Through analysis, 16 signs and symptoms in six categories were used to distinguish IIM cases from the comparison cases. The following table defines the signs and symptoms:
| Sign or Symptom | Definition |
| Age of onset | |
| Age of onset of first symptom assumed to be related to the disease ≥18 years and <40 years | 18≤Age (years) at onset of first symptom assumed to be related to the disease <40 |
| Age of onset of first symptom assumed to be related to the disease ≥40 years | Age (years) at onset of first symptom assumed to be related to the disease ≥40 |
| Muscle weakness | |
| Objective symmetric weakness, usually progressive, of the proximal upper extremities | Weakness of proximal upper extremities as defined by manual muscle testing or other objective strength testing, which is present on both sides and is usually progressive over time |
| Objective symmetric weakness, usually progressive, of the proximal lower extremities | Weakness of proximal lower extremities as defined by manual muscle testing or other objective strength testing, which is present on both sides and is usually progressive over time |
| Neck flexors are relatively weaker than neck extensors | Muscle grades for neck flexors are relatively lower than neck extensors as defined by manual muscle testing or other objective strength testing |
| In the legs, proximal muscles are relatively weaker than distal muscles | Muscle grades for proximal muscles in the legs are relatively lower than distal muscles in the legs as defined by manual muscle testing or other objective strength testing |
| Skin manifestations | |
| Heliotrope rash | Purple, lilac-coloured or erythematous patches over the eyelids or in a periorbital distribution, often associated with periorbital oedema |
| Gottron’s papules | Erythematous to violaceous papules over the extensor surfaces of joints, which are sometimes scaly. May occur over the finger joints, elbows, knees, malleoli and toes |
| Gottron’s sign | Erythematous to violaceous macules over the extensor surfaces of joints, which are not palpable |
| Other clinical manifestations | |
| Dysphagia or oesophageal dysmotility | Difficulty in swallowing or objective evidence of abnormal motility of the esophagus |
| Laboratory measurements | |
| Anti-Jo-1 (anti-histidyl-tRNA synthetase) autoantibody present | Autoantibody testing in serum performed with standardized and validated test, showing positive result |
| Elevated serum levels of creatine kinase (CK)* or lactate dehydrogenase (LD)* or aspartate aminotransferase (ASAT/AST/SGOT)* or alanine aminotransferase (ALAT/ALT/SGPT)* | The most abnormal test values during the disease course (highest absolute level of enzyme) above the relevant upper limit of normal |
| Muscle biopsy features—presence of: | |
| Endomysial infiltration of mononuclear cells surrounding, but not invading, myofibres | Muscle biopsy reveals endomysial mononuclear cells abutting the sarcolemma of otherwise healthy, non-necrotic muscle fibres, but there is no clear invasion of the muscle fibres |
| Perimysial and/or perivascular infiltration of mononuclear cells | Mononuclear cells are located in the perimysium and/or located around blood vessels (in either perimysial or endomysial vessels) |
| Perifascicular atrophy | Muscle biopsy reveals several rows of muscle fibres, which are smaller in the perifascicular region than fibres more centrally located |
| Rimmed vacuoles | Rimmed vacuoles are bluish by H&E staining and reddish by modified Gomori trichrome stains |
Cut-off probabilities based on the cumulative value of score points were determined to define possible, probable and definite IIM. The researchers further analyzed the score point values and probabilities based on whether a muscle biopsy was available. The probabilities were determined as follows, which balanced both sensitivity and specificity for an optimal result:
- Less than 50% – Not IIM
- Greater than or equal to 50% but less than 55% – Possible IIM
- Greater than or equal to 55% but less than 90% – Probable IIM
- Greater than or equal to 90% – Definite IIM
Then, if a patient falls into the Probable or Definite IIM category, a classification tree was used to further classify IIM into subgroups based on signs and symptoms, with the exception of immune-mediated necrotizing myopathy (IMNM), juvenile polymyositis and hypomyopathic DM, because the sample size for these cases was too small to be meaningful.
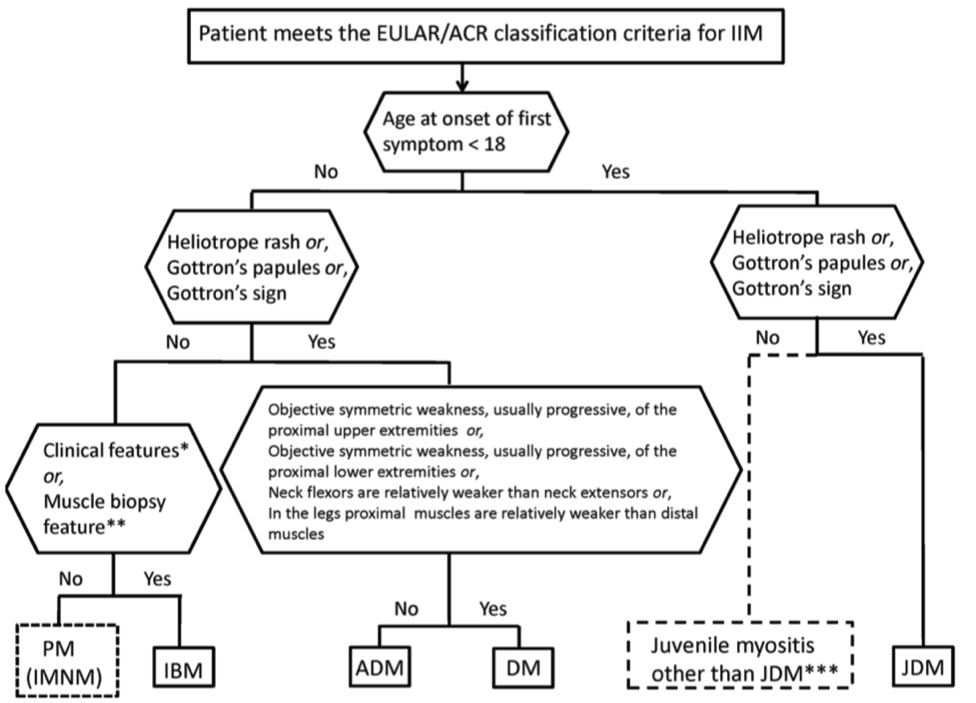 The authors included additional elements to the development of the classification system that are worth noting. For patients with juvenile dermatomyositis and the presence of skin rashes, the scoring system does not include muscle biopsies, which were not frequently available. The criteria correctly classified 97% of juvenile dermatomyositis (DM) patients without muscle biopsy data. The authors also compared their results with other classification systems. Some were found to have either high sensitivity and low specificity (such as Bohan and Peter), or high specificity and low sensitivity. The study also included multiple validation tests that confirmed excellent performance of the criteria.
The authors included additional elements to the development of the classification system that are worth noting. For patients with juvenile dermatomyositis and the presence of skin rashes, the scoring system does not include muscle biopsies, which were not frequently available. The criteria correctly classified 97% of juvenile dermatomyositis (DM) patients without muscle biopsy data. The authors also compared their results with other classification systems. Some were found to have either high sensitivity and low specificity (such as Bohan and Peter), or high specificity and low sensitivity. The study also included multiple validation tests that confirmed excellent performance of the criteria.
The result of this study is that new classification criteria for IIM have been developed that are:
- more accurate than previous criteria in identifying IIM subgroups
- based on a relatively small number of criteria that are clearly defined
- have high sensitivity and specificity
- and have been at least partly validated
These criteria can be used as diagnostic and classification criteria for clinical trials. One of the limitations of this study is that the majority of the cases were Caucasian patients and could differentiate among ethnic groups, if differences exist. Another limitation was that subjects had to have the disease for 6 months and did not include new onset patients. Three categories did not include enough test subjects, despite having taken 10 years to gather the data. And there was a very low frequency of autoantibody data in the study. Only the anti-Jo-1 autoantibody had enough cases to include in the criteria. This set of criteria should continue to be updated and refined as new methods of testing and detecting IIM become available.
The authors also created a web calculator based on the criteria, which can be found at the following link: www.imm.ki.se/biostatistics/calculators/iim
Read Full Article Tags: dermatomyositis diagnosing myositis inclusion body myositis juvenile myositis myositis necrotizing autoimmune myopathy polymyositis




